





Мукополисахаридоз
Мукополисахаридозы présentent un grand groupe des maladies monogénétiques héréditaires résultant  de la violation dans les lysosomes du procès à plusieurs degrés de la catalyse enzymatique гликозаминогликанов (des EIDERS).
de la violation dans les lysosomes du procès à plusieurs degrés de la catalyse enzymatique гликозаминогликанов (des EIDERS).
Гликозаминогликаны sont complexe de l'hétérosucre, qui comprennent de полисахаридных des chaînes comprenant les restes сульфатированного гексозамина et глюкуроновой les acides. Дерматансульфат, кератансульфат, гепарансульфат, khondroitin-6 et le khondroitin-4-sulfate sont гликозаминогликанами.
Pour la première fois la maladie мукополисахаридоз était décrite en 1917 par Gourlerom. À présent on sait près de 10 aspects génétiques мукополисахаридоза, cinq de qui résultent de la violation de l'activité сульфатаз, quatre – гликозидаз et un type se développe au manque трансферазы. À мукополисахаридозе il y a une accumulation des particules dans l'organisme que conduit à l'apparition à la personne des divers symptômes et les pathologies.
La maladie est héritée selon aoutosomno-retsessivnomou au type.
Que se passe à мукополисахаридозе ?
En présence de la maladie il y a une défaite du système лизосомных des ferments, qui prennent part au catabolisme гликозаминогликанов. À la suite du déficit des ferments dans les organismes et les tissus il y a une accumulation гликозаминогликанов que permet de ranger мукополисахаридоз la maladie de l'accumulation.
L'accumulation гликозаминогликанов amène à la violation de l'état fonctionnel des divers systèmes et les organismes, et puisque гликозаминогликаны font partie du tissu conjonctif, un de principaux symptômes мукополисахаридоза est la défaite systémique du squelette et le retard du développement physique. Particulièrement c'est exprimé à I, IV, VI types мукополисахаридоза.
Les symptômes мукополисахаридоза
Aux symptômes мукополисахаридоза se rapporte le retard de la croissance, qui commence à la fin de la première année de la vie de l'enfant. Il faut marquer les traits du visage grossiers : une grande langue surplombant le front, la déformation des dents et les oreilles, гипертелоризм. On déforme le thorax, est vivement exprimé кифоз des services lombaires et de poitrine de l'épine dorsale. Sont caractéristiques гепатоспленомегалия, inguinal et les hernies ombilicales, la restriction de la mobilité des articulations.
Sur l'examen radiographique on peut voir "de poisson" позвонки, l'ossification précoce de la couture zatylotchno-sincipitale. De plus la formation des noyaux de l'ossification n'est pas violée.
Aux symptômes neurologiques мукополисахаридоза se rapportent total moteur заторможенность, 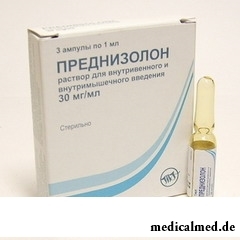 l'hypotension diffusive musculaire. L'affaiblissement de la rumeur et la réduction de l'intelligence est aussi caractéristique pour de différents types мукополисахаридоза.
l'hypotension diffusive musculaire. L'affaiblissement de la rumeur et la réduction de l'intelligence est aussi caractéristique pour de différents types мукополисахаридоза.
Les types мукополисахаридоза
Selon le degré выраженности des symptômes psychologiques et les changements osseux, ainsi que selon la vitesse de la progression des violations de change on sait 7 types мукополисахаридоза :
- I type, qu'appellent aussi le syndrome de Gourlera. À la maladie le développement d'une manière caractéristique rapide. Dans l'urine des patients il y a un grand contenu хондроитинсульфата à et гепаритинсульфата. Est hérité selon aoutoimmounno-retsessivnomou au type.
- II type, ou le syndrome de Gountera. À la maladie donnée s'enregistre de pigment ретинит, la disacousie. La maladie progresse lentement. Dans l'urine on observe aussi le contenu augmenté гепаритинсульфата et хондроитинсульфата à, mais en plus petites quantités. Est hérité selon рецессивному au type, dépend du plancher.
- III type, le syndrome de Sanfilippo. Pour le type donné la débilité d'une manière caractéristique lourde et une grande quantité гепаритинсульфата dans l'urine. La succession aoutoimmounno-retsessivnoe.
- IV type, ou le syndrome de Morkio. On marque la déformation essentielle du squelette, particulièrement le thorax. À la différence d'autres types l'absence du trouble de la corné, la réduction de l'intelligence et l'apparition des traits du visage grossiers est caractéristique.
- V type, le syndrome de Cheje. Se caractérise par le trouble de la corné et la déformation modérément-exprimée du squelette. Est hérité selon aoutoimmounno-retsessivnomou au type.
- VI type, ou le syndrome de Maroto-Lami. Sont caractéristique le retard dans la croissance, бочкообразная le thorax, les traits du visage grossiers.
- VII type apparaissant en conséquence du manque bêta-глюкуронидазы.
Le diagnostic мукополисахаридоза
Le diagnostic de la maladie se fonde sur la tenue des études généalogiques, cliniques et biochimiques.
Le traitement мукополисахаридоза
La maladie a principalement le traitement symptomatique. Au patient fixent les préparations hormonales : преднизолон, тиреоидин, АКТГ pour la répression de la production мукополисахаридов. Au traitement мукополисахаридозов au patient fixent grand дозировки de la vitamine A et les préparations pour la normalisation de l'activité cordiale. Peuvent fixer en cas de nécessité les moyens cytostatiques.
La prophylaxie мукополисахаридоза
La prophylaxie spécifique de la maladie n'existe pas. La tenue пренатальной du diagnostic pour la révélation du manque des ferments dans les carreaux amniotiques est nécessaire.
Selon les études, les femmes buvant quelques verres de la bière ou la faute par semaine, ont le risque augmenté de tomber malade du cancer de la poitrine.

Que n'inventeront pas seulement souhaitant avoir une belle figure. Voici la dernière innovation – pour похудения il faut manger la nourriture grasse. ра...
Le paragraphe : la Séance de projection
Plusieurs parents des enfants à l'âge de 2-4 ans se heurtent avec excessivement par la conduite capricieuse de l'enfant. Le petit épuise aux constantes nous pleurons par les caprices non seulement les parents, mais aussi. Dans quoi de la raison des caprices d'enfant. Et comment avec eux lutter ?...
Le paragraphe : la Séance de projection
L'hémorragie est une des maladies les plus répandues de la personne, chaque année dans le monde est enregistré près de 6 millions de cas de cette pathologie. D'après les données de la statistique médicale, les hémorragies se passent presque à trois fois plus souvent, que les infarctus du myocarde. La maladie se rapporte vers lourd, et a le bilan décevant : la létalité arrive jusqu'à 40 % parmi les femmes et 25 % parmi les hommes. La partie considérable des patients qui ont éprouvé l'hémorragie et ne peut pas entièrement se rétablir. Nous proposons aux lecteurs de prendre connaissance...
Le paragraphe : les Articles sur la santé
La sclérotique et la membrane muqueuse de l'oeil sont d'une manière intense approvisionnée en vaisseaux sanguins, la tâche de qui - saturer les tissus nerveux de l'organisme питате...
Le paragraphe : les Articles sur la santé
Аюрведа - le plus ancien целительская la pratique nous venant vers de l'Inde. Elle représente la doctrine sur le maintien de la santé physique, psychologique et morale de la personne avec l'aide de l'ensemble des procédures insérant le régime, le nettoyage de l'organisme, respiratoire упр...
Le paragraphe : les Articles sur la santé
Contrairement à l'opinion répandue, la sclérose dispersée (РС) n'est pas liée ni aux changements scléreux des murs des récipients, ni avec l'oubli d'âge et les problèmes avec la concentration de l'attention. Cette maladie a аутоиммунную la nature. Le procès pathologique s'exprime dans la dégradation du tissu nerveux et la destruction de sa couche extérieure - la myéline. Par le résultat du développement de la maladie il y a des défaites multiples du système cérébro-spinal, qui se manifestent par la réduction de la vue, la fatigue rapide, sur...
Le paragraphe : les Articles sur la santé
Avec la grippe à nous, malheureusement, il faut se heurter presque chaque année. Apparemment, la maladie si fréquente doit être изуче...
Le paragraphe : les Articles sur la santé
Chacun de nous remarquait plus d'une fois que les gens ayant le même âge de passeport, ne sont pas du tout semblables parfois pour des monoannées. Un à l'âge de 40-45 ans a l'air déjà presque du vieillard, et l'autre et à 60 est jeune, énergique et complet les vies. Le fait est que l'état нашег...
Le paragraphe : les Articles sur la santé
Le café – la boisson aimée de plusieurs. Pour ses dernières décennies bien des fois annonçaient déjà cela très nuisible, extraordinairement utile et même nécessaire à l'activité vitale normale. Malgré le fait que ce produit devienne depuis longtemps pour nous habituel, il y a beaucoup de mythes sur les propriétés du café et son influence sur l'organisme de la personne. Avec les plus répandu des erreurs semblables les lecteurs peuvent faire connaissance aujourd'hui....
Le paragraphe : les Articles sur la santé
Aiment chanter tout. De petits enfants s'occupent avec plaisir du chant, non en réfléchissant spécialement à l'atteinte à la mélodie. Les adultes le plus souvent...
Le paragraphe : les Articles sur la santé
L'épilepsie est une des maladies répandues neurologiques. Aux parents, quels enfants souffrent de cette maladie, il faut se heurter aux rumeurs et les erreurs, plusieurs de qui se sont gardés dès les temps du moyen âge....
Le paragraphe : les Articles sur la santé
Non, probablement, la personne, qui ne serait pas malade du refroidissement. Le rhume, la toux, le mal de tête – on savent ces symptômes à chacun. Le pic des maladies de refroidissement vient pour l'automne. ОРВИ sont venus déjà aux écoles et les jardins d'enfants, la grippe pénètre lentement dans les villes, bref, l'hiver tout près!...
Le paragraphe : les Articles sur la santé
Avoir l'air sain et soigné signifie non seulement plaire aux proches, mais aussi se sentir fort, assuré et ayant lieu. Спе...
Le paragraphe : les Articles sur la santé
L'églantier – une des plantes les plus répandues décoratives et médicinales, croissant pratiquement sur tout le territoire de notre pays. À la plupart des Russes c'est un beau buisson est connu, avant tout, comme la source des fruits extraordinairement riches en les vitamines....
Le paragraphe : les Articles sur la santé
Les yeux – un des domaines les plus vulnérables sur la personne, c'est pourquoi les changements d'âge les concernent en premier lieu. Si on peut garder la jeunesse du regard pour des longues années, et quelles procédures proposent pour l'acquisition de ce but les cosmétologues ? Et peut être, la seule variante du rajeunissement est l'opération chirurgicale – блефаропластика ? Nous essaierons comprendre cette question....
Le paragraphe : les Articles sur la santé
L'été de cette année en Russie s'est présenté très ambigu. Les régions souffraient cela de la chaleur impitoyable, des pluies battantes, temps...
Le paragraphe : les Articles sur la santé
Par tout le slogan connu «Ménagez les hommes!» Est apparu non à la place vide. Au sens défini, la nature a créé les représentants du sexe fort beaucoup moins adapté vers les désarrois de vie, que cela semble à première vue. Selon la statistique, les hommes sont malade plus souvent...
Le paragraphe : les Articles sur la santé
Le corps de la personne presque sur 60 % comprend l'eau. C'est tellement important pour le fonctionnement normal de l'organisme que la perte seulement une et demi pour-cent du liquide amène déjà aux conséquences les plus désagréables. Les problèmes liés au déficit de l'eau, peuvent настигнуть et la personne la plus saine, s'il, par exemple, passe quelques heures sous le soleil ardent, n'ayant pas pris la boisson, mais ajuster l'état de santé dans ce cas il est très simple. Il est beaucoup plus complexe minimiser les conséquences des autres raisons sur...
Le paragraphe : les Articles sur la santé
On peut appeler les antibiotiques - les combinaisons chimiques réprimant la croissance des bactéries - comme la rupture dans le domaine de la médecine, permettant de sauver чел...
Le paragraphe : les Articles sur la santé
Sur l'influence des jours de déchargement sur l'organisme est dit beaucoup – sur les dignités, ainsi que sur les manques. Il Croit que le jour de déchargement en forme du monorégime à court terme est utile, en contribuant au retrait effectif des scories de l'organisme, tandis qu'irrégulier, trop п...
Le paragraphe : les Articles sur la santé
Фобия – la peur importune du contenu défini manifestée dans la situation concrète à contrecoeur de la personne. Les notions фобии et la peur sont pareilles, cependant si la peur est la fonction naturelle protectrice de la mentalité, фобия est son rejet. Ainsi la personne peut avoir la peur inconsciente sans raison accompagnée par les symptômes névrotiques (la sudation excessive, le tremblement, les frissons) devant quelque phénomène ordinaire – par exemple, le voyage en métro ou le chien simple....
Le paragraphe : les Articles sur la santé
Les préparations cessant ou opprimant l'activité vitale des microorganismes pathogènes, sont appliquées largement dans la pratique clinique avec 4...
Le paragraphe : les Articles sur la santé
Arrive la saison des congés. Plusieurs Russes rêvent déjà du repos sur la nature, les voyages touristiques, de belles plages maritimes. À cette époque on ne veut pas penser des problèmes avec la santé et d'autres objets désagréables, il y a cependant des sujets, qui demandent l'attention. L'été...
Le paragraphe : les Articles sur la santé
L'opinion univoque sur la raison directe de l'apparition du cancer de la peau chez les savants est absente aujourd'hui. On établit exactement seulement les facteurs contribuant au développement de cette maladie. À ceux-ci se rapportent : l'influence de longue durée sur la peau des rayons ultraviolets, l'irradiation radioactive, les traumas thermiques, l'endommagement de la peau par les substances agressives chimiques (les goudrons, les acides, щелочами etc.), la prédisposition héréditaire (la présence des néoplasmes malins de la peau dans l'anamnèse familiale), à...
Le paragraphe : les Articles sur la santé
Ce n'est pas un secret pour personne que notre pays est un des plus "buvant" au monde. À la compréhension claire de ce que l'utilisation des solides...
Le paragraphe : les Articles sur la santé
La quantité долгожителей est très pas grande. Une personne vit jusqu'à l'âge de 90 années de 5 mille, et la frontière centenaire enjambe seulement un de 20 mille. D'ailleurs, les médecins affirment que chacun de nous tout à fait dans l'état d'influencer le destin personnel. De plus les paroles иде...
Le paragraphe : les Articles sur la santé
Selon la statistique 25-30 % des femmes jusqu'à 40 ans sont malade de la cystite. Avec l'âge ce paramètre augmente, en plus plusieurs ne se trouvent pas dans la statistique, parce que ne s'adressent pas au médecin. Le plus triste qu'après les visites régulières chez les médecins, l'accueil de longue durée des antibiotiques et la vie en régime «il faut se ménager» la cystite revient en tout cas à la moitié des femmes. Les symptômes de la cystite sont univoques et les confondre avec rien on ne peut pas : la douleur dans la vessie, le pyrosis à l'urination, les envies fréquentes aller à la toilette, вынужда...
Le paragraphe : les Articles sur la santé
Selon la statistique, personne souffre des pathologies de la glande thyroïde dans le monde plus de 500 millions. Les défaillances dans le travail de cet organisme conduisent vers est lourd...
Le paragraphe : les Articles sur la santé